Background on Type 2 Diabetes
Type 2 diabetes is broadly defined as a combination of insulin resistance and beta-cell dysfunction, where insulin signalling and production becomes insufficient. Before reaching the stage of type 2 diabetes, one can experience insulin resistance for years, until the pancreatic function deteriorates to the point of meeting the diagnostic criteria for diabetes.
In this post, we will explore how an increased amount of lipids/fats blocks several steps in the insulin signaling cascade, contributing to insulin resistance. We’ll also examine how reductive stress and increased fatty acid oxidation inhibit insulin release from the pancreatic beta-cells.
Normal Insulin Release in Beta Cells:
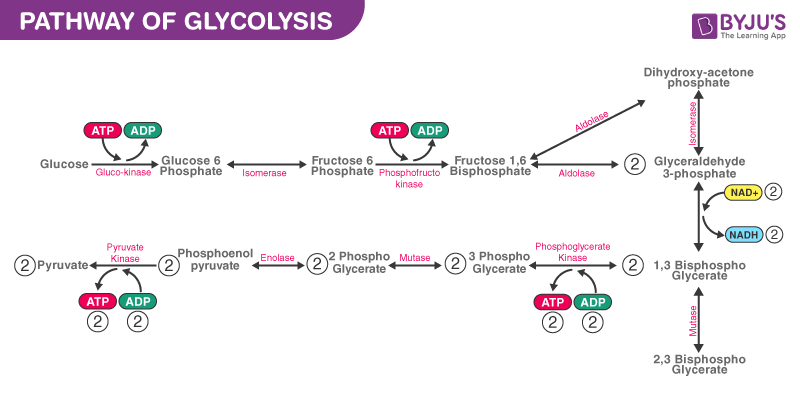
Insulin release in response to rising plasma glucose levels begins when glucose enters beta cells in the pancreas through GLUT2 receptors, independent of insulin. This leads to an increase in cellular energy through glycolysis, the Krebs cycle, and the electron transport chain, raising the ATP/ADP ratio. This increase inhibits a potassium-ATP channel, reducing potassium efflux and depolarizes the beta cell. As a result, a voltage-dependent calcium channel opens, causing calcium influx, which triggers the exocytosis of insulin via microtubule-mediated processes.
(Reference: Koeppen BM, Stanton BA. Berne & Levy Physiology. 7th ed: Elsevier; 2018.)

Insulin Action:
Insulin travels through the bloodstream to tissues, where it activates insulin receptors, primarily in muscles and fat tissue. Activation of these receptors initiates a dephosphorylation cascade, which leads to activation of several signaling pathways.
Insulin promotes the following:
- Increased glucose uptake by sending GLUT4 vesicles to the cell membrane.
- Stimulation of glycolysis through the increased concentration of intracellular glucose.
- Activation of pyruvate dehydrogenase, which boosts cellular energy production via the Krebs cycle.
- Stimulation of glycogen synthesis (glycogenesis), replenishing muscle sugar stores.
- Activation of de novo lipogenesis, especially if glucose is not immediately used or if there is reduced electron flow and there by a decreased amount of electron acceptors downstream of glycolysis.
- Activation of the mTOR signaling pathway, enhancing protein synthesis and reducing protein breakdown.
- Inhibition of gluconeogenesis and VLDL export, reducing new sugar production (a hallmark of diabetes is increased gluconeogenesis) and decreasing fat export from the liver. This reduces fat burning in tissues.
- Upregulation of glycolytic enzyme transcription and mitogenic factors, leading to increased cell division and anabolic effects.
(Reference: Koeppen BM, Stanton BA. Berne & Levy Physiology. 7th ed: Elsevier; 2018.)
Dysregulated Mechanisms in Diabetes
In type 2 diabetes, the exocytosis of insulin is impaired, resulting in lower insulin release. This is due to a failure in the link between glycolysis, the Krebs cycle, and the electron transport chain, which leads to insufficient ATP production. Insulin signaling is also impaired, which is known as insulin resistance.
Redox Imbalance and Glucose Metabolism Blockage in Beta Cells
Recent research suggests that beta-cell dysfunction in type 2 diabetes is not primarily due to apoptosis (programmed cell death) but rather reduced coupling between glucose metabolism and insulin release. Although apoptosis may occur, it is insufficient to explain the substantial reduction in insulin secretion seen in type 2 diabetes.
A key factor in this dysfunction is the increased metabolism of fatty acids through the Randle cycle, which suppresses glucose metabolism be outcompeting glucose metabolism for intermediates substrates.
Fat deposits in the pancreas are also linked to diabetes development, and one could think that the accumulation of fat makes a higher local concetration of fats, inhibiting glucose metabolism.
Increased fatty acid oxidation causes a relative increase in FADH2 compared to NADH, leading to a “clogging” of complex I in the electron transport chain. This results in a decreased NAD+/NADH ratio, causing reductive stress, becuase the reduced electron carriers have nowhere to deliver the excess electrons. This reductive stress, in turn, leads to oxidative stress and the accumulation of reactive oxygen species (ROS). ROS accumulation can further impair glucose metabolism, by inactivation of glyceraldehyde 3-phosphate dehydrogenase, a key glycolytic enzyme, and thereby blocking the glycolytic pathway, and ultimately, reducing insulin release in the pancreatic beta cells.
(References: Haythorne E, et al. Nature Communications. 2019; Yan LJ. J Diabetes Res. 2014.)
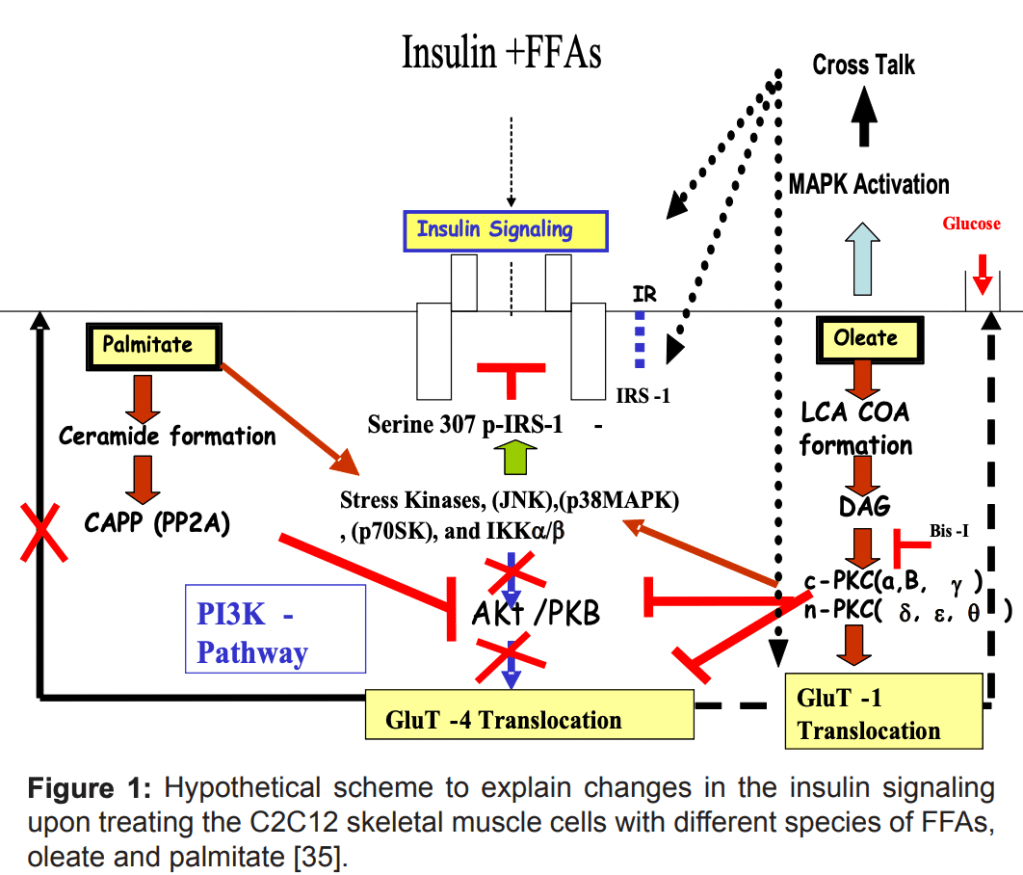
Insulin Resistance Driven by (PUFA) Fatty Acid Infusions
Insulin resistance in tissues is also influenced by fatty acid oxidation. Lipids and fatty acids contribute to insulin resistance through several pathways, especially via inflammatory cascades. One key pathway is the activation of NF-kappa-beta signaling. Animal studies have shown that inhibition of this pathway, either genetically or through anti-inflammatory drugs like aspirin, prevents lipid-induced insulin resistance.
Elevated Fatty acids in the bloodstream activate the NF-kappa-beta pathway, which inhibits glucose transport (GLUT4) and phosphorylation, resulting in reduced glycolysis. This is not necessarily related to the classical understanding of the Randle cycle (competition between substrates) but rather to lipid-mediated inflammation, which disrupts insulin signaling.
Fatty acid infusions, and in one study, particularly PUFA (as used in the rat study with Liposyn II), induces insulin resistance by activating inflammatory pathways and blocking insulin signaling.
(References: Boden G, et al. Diabetes. 2005.)
Fatty Acid Accumulation and Insulin Resistance
Besides from activation of the inflammatory pathways, fatty acid accumulation also activates Protein Kinase C (PKC), which inhibits insulin signaling by phosphorylating insulin receptor substrates like IRS-1. Both saturated fats (e.g., palmitate) and monounsaturated fats (Oleate/MUFAs) can also induce insulin resistance, although through different mechanisms.
While the Saturated fat Palmitate has a mechanistic blockage through PKB inhibition, the oleate MUFA has more of an inhibiting effect on the phosphorylation of PKB – And by not activating PKB, the signal does not go further downstream, and the GLUT4 glucose transporter is not sufficiently transported to the cellular membrane.
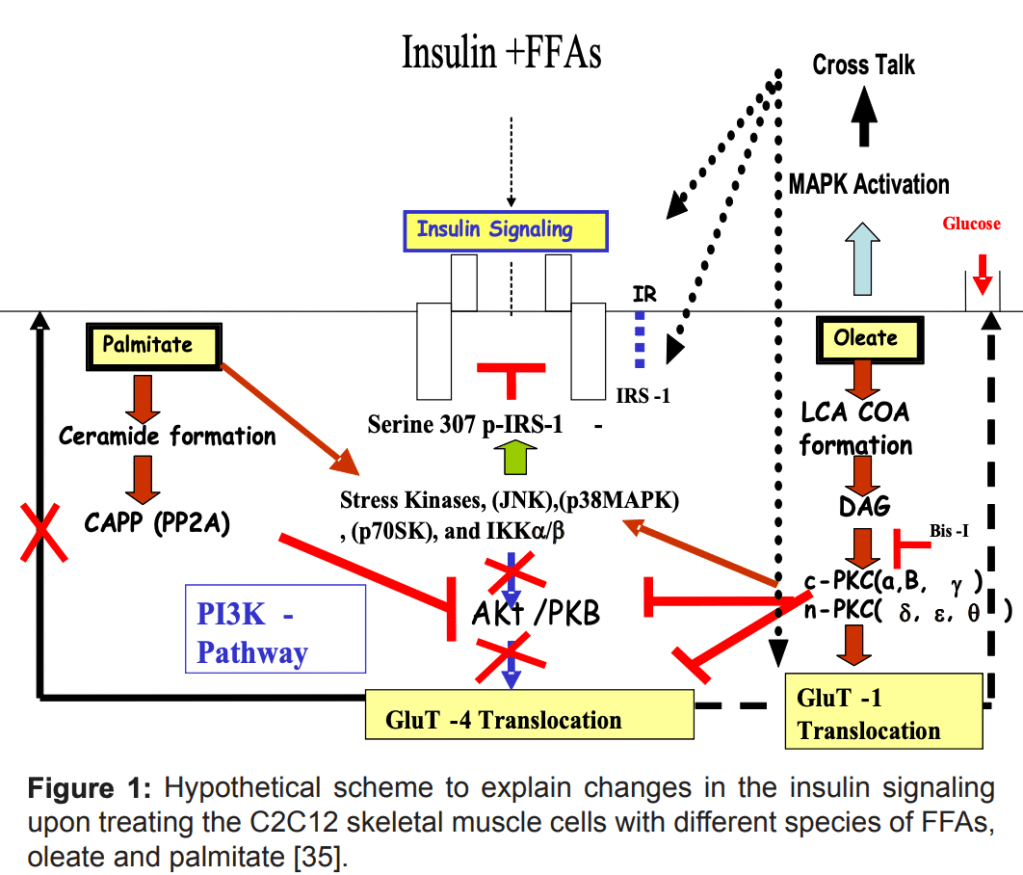
This lipid-induced insulin resistance can occur even in healthy individuals within hours of lipid infusion.
(Muoio, D. M., & Newgard, C. B. (2008). Mechanisms of fatty acid-induced insulin resistance in muscle and liver. Journal of Diabetes & Metabolism, 2155-6156.)
Summary:
- Lipids block insulin signaling, probably through inflammation via the NF-kappa-beta pathway, which ends up phosphorylating the IRS-1 ( essentially blocking the phosphorylation cascade when it is already phosphorylated), but also through lipid blockage of AKT/ PKB further down stream in the signalling cascade.
- Aspirin, or inhibition of the NF-Kappa-Beta inflammation, can reverse insulin resistance caused by lipid infusions.
- Reductive stress from excess fatty acid oxidation blocks glucose metabolism and insulin release.
What Can Be Done to Address Diabetes and Insulin Resistance?
To combat diabetes, it’s essential to:
- Reduce reductive stress (excess electron accumulation).
- Improve insulin signaling and enhance glucose metabolism while lowering fatty acid metabolism.
It may be beneficial to:
- Avoid prolonged fasting and stress, which floods the system with accumulated fats.
- Reduce fat intake, particularly polyunsaturated fats (PUFAs), which have been shown to reduce the NAD+/NADH ratio and increase reductive stress.
Finally, increasing the NAD+ pool through Vitamin B3 supplementation could help restore balance in the redox state and improve glucose metabolism.
(References: Yan LJ, et al. 2014; Haythorne E, et al. 2019.)


Leave a comment