Lidt baggrund for type 2 diabetes.
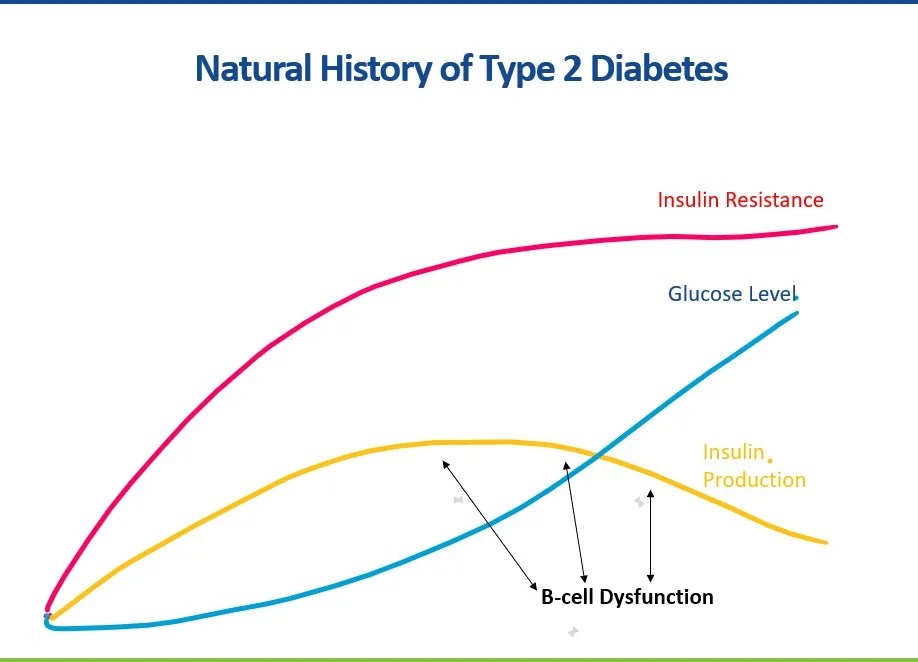
Type 2 diabetes defineres i korte træk som en blanding af insulinresistens og beta celle dysfunktion, hvorved insulinproduktionen ikke er tilstrækkelig. Inden man når til stadiet der hedder type 2 diabetes, kan man have insulinresistens i årevis op til at pancreas funktionen forringes i en sådan grad at man kan opfylde de diagnostiske kriterier for diabetes
Vi skal i dette indlæg se på hvordan en øget mængde lipider/fedtstoffer blokerer flere skridt i insulinsignalleringskaskaden og medvirker til insulinresistensen, og så skal vi se på hvordan reduktiv stress og øget fedtsyre oxidation hæmmer insulinudløsningen fra pancreas.
Først skal vi kigge på de almindelige funktioner i kroppen, før vi kan forstå de syge processer.
Normal insulin frigivelse i betaceller:
Den normale insulinstigning i plasma, induceres af øget glukose i blodet, som via glut2 receptorerer entrerer betacellerne i pancreas uafhængigt af insulin.
Når betacellen modtager glukose, så stiger energiomsætningen via glykolysen, krebs cyklus og elektrontransportkæden – hvilket medfører en stigning i ATP/ADP ratioen, hvilket hæmmer en Kalium-ATP kanal, således at kalium efflux/udadtransport hæmmes, således at der sker en depolarisering af betacellen, hvilket aktiverer en spændingsafhængig calciumkanal. Calcium influx i cellen stiger, og der sker så en mikrotubuli medieret exocytose af insulin – (s. 705 – Koeppen BM, Stanton BA. Berne & Levy Physiology. 7th ed: Elsevier; 2018.)
Insulin virkning:
Insulin kommer med ud til vævet via blodet, hvor det aktiverer insulinreceptoren i det væv hvor der er insulinafhængig glukose optag, som f.eks. i den hvilende muskel eller i fedtvævet.
Insulinrecopteren aktiveres, og det sætter en kaskade af defosforylering i gang, der aktiverer flere signalleringsveje.
Insulin virker således ved at:
– Øge cellens glukose optag, ved at sende glut 4 vesikler til membranen.
-Glykolysen stimuleres – bl.a. via den øgede koncentration af glukose i cellen.
-Pyruvatdehydrogenasen, det begrænsende led mellem glykolysen og krebscyklus stiger – den cellulære elektrontransport og energi dannelse forbedres altså.
-Glukogenesen stimuleres – nydannelse af glukogen/muskelsukker således at depotet af muskelsukker genopbygges efter brug.
– De novo lipogenesen aktiveres, især hvis glukosen ikke bliver brugt i den grad det kommer ind, eller hvis der af en eller anden årsag er et nedsat elektronflow downstream for glukolysen.
– MTOR signalvejen aktiveres, og der sker en øgning i proteinsyntese og falde i protein nedbrydning.
– Glukoneogenesen nedreguleres, og VLDL eksport nedreguleres. Dvs. der laves ikke ny sukker, som jo er et af kendetegnende ved diabetes, at glukoneogenesen er opreguleret. Samtidig eksporteres der ikke fedt i form af VLDL i samme grad til blodet fra leveren, og fedtforbrændingen i vævet sænkes altså.
– Transkription af glykolytiske enzymer stiger, og der sker en øgning i mitogene faktorer således at celledeling opreguleres. Således har insulin også en anabolsk natur i form af øget celledeling og transkriptorisk opregulering af glykolytiske enzymer.
(s. 706-708 – Koeppen BM, Stanton BA. Berne & Levy Physiology. 7th ed: Elsevier; 2018.)
Dysregulerede mekanismer
Nu har vi været lidt inde over den normale funktion af insulin og hvordan insulin frigives fra beta cellerne i pankreas.
Nu skal vi kigge på hvordan disse mekanismer er dysreguleret i diabetes.
I diabetes type 2 virker exocytosen af insulin ringere således at der udløses mindre insulin, fordi der er et eller andet i leddet mellem glykolysen, krebs cyklus og elektrontransportkæden der ikke fører til en sufficient ATP stigning.
Samtidig virker insulinsignalleringen ringere, hvilket vi kalder insulinresistens.
Redox ubalance, reduktiv stress og blokering af glukosemetabolisme i betaceller.
I en artikel i nature, er der noget der peger i retning af at det ikke er apoptose (Programmeret celledød) af beta celler der forårsager betacelle dysfunktion, men nærmere en nedsat coupling mellem glukose metabolismen og exocytosen/frigivelsen af insulin.
Så selvom der måtte være en grad af apoptose, så er det ikke nok til at kunne stå for den grad af nedsat insulinfrisætning der er i type 2 diabetes.
Haythorne E, Rohm M, van de Bunt M, Brereton MF, Tarasov AI, Blacker TS, et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. Nature Communications. 2019;10(1):2474.
Vi ved jo at noget af det der hæmmer glukosemetabolismen er en øget fedtsyre metabolisme gennem randle cycle. Vi ved også at en af de helt store risici for diabetes udvikling er fedtaflejringer på pancreas –
https://pubmed.ncbi.nlm.nih.gov/34571257/
Men generelt har vi at gøre med en sænkning i raten af ATP produktion, og det kan altså være en grad af reduktiv stress i redox balancen for elektrontransport fra maden til ATP – Altså en overflod af NADH relativt til NAD+, således at der en ophobning af elektroner i energikæden, fordi der bl.a. gennem fedtsyre oxidation sker en reduktiv stress ved at NADH strander til ved elekrontransportkædens komplex 1.
Denne ophobning af elektroner fører så samtidig til en oxidativ stress ved dannelse af frie radikaler/ROS.
Når NADH og ROS ophobes, kan Glyceraldehyd 3-fosfat dehydrogenase (GADPH) inaktiveres, og det resulterer i en blokade af den glykolytiske signalvej, resulterende i en ophobning af glycerol 3-phosphat og metabolitterne opstrøms for denne, herunder glukose.
med andre ord, falder evnen til at bruge glukose og sætte gang i insulinfrigivelsen som følge heraf. pga. en ophobning af elektroner i mitokondrierne.
Glykolysen har altså brug for NAD+, den oxiderede form af Nicotinamide adenin dinucleotid, og hæmmes hvis ikke der er tilstrækkeligt NAD+.
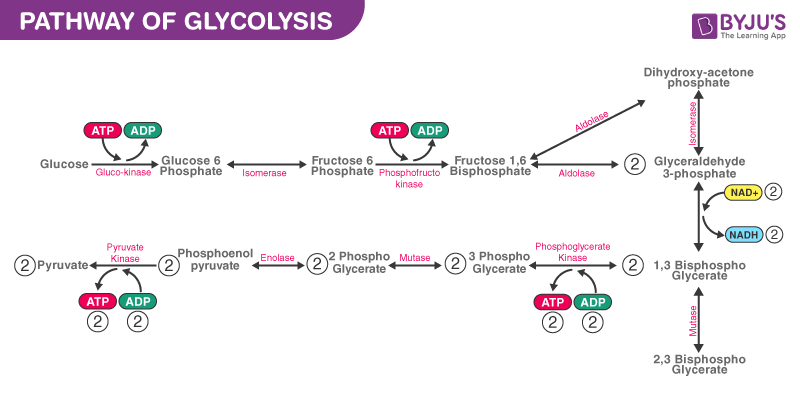
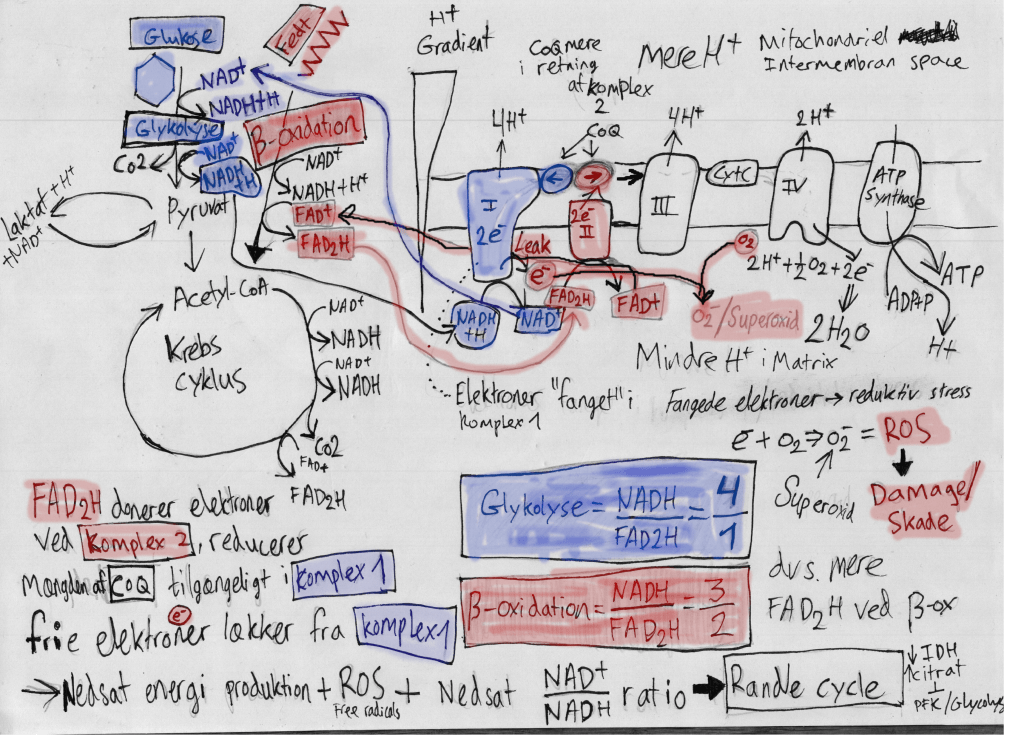
Så langt så godt – Fedtsyre oxidation medfører en relativ øgning FADH2 relativt til NADH, førende til en “clogging” ved kompleks 1 i elektrontransportkæden, hvilket fører til et nedsat NAD+/NADH ratio – altså reduktiv stress, som fører til oxidativ stress/ROS og en ophobning af glukose opstrøms, med yderligere ROS og celleskade til følge.
Insulinresistens som følge af fedtsyre infusioner:
Hvis vi kigger på vævets insulinresistens, så er fedtsyre oxidation også på spil. Ved insulinresistens, har vævet en nedsat evne til at respondere på insulinsignalet hvor led i kaskaden hæmmes.
Noget tyder på at lipider/fedtsyrer igen er på spil, og medierer flere veje der fører til insulinresistens – den vigtigste relateret til inflammationskaskaden.
Der sker en stigning i inflammationskaskaden via NF-kapppa-beta signalvejen – mus behandlet med høje doser af Aspirin/acetylsalicylsyre, som er antiinflammatorisk og som uspecifikt hæmmer NF-kappa-beta signalvejen, eller mus der genetisk har fået knockoutet NF-kappa-beta signalleringsvejen, får ingen insulinresistens i forbindelse med lipidinfusioner.
Noget tyder altså på at en lipidmedieret inflammation/NF-kappa-beta aktivering er medvirkende til insulinresistens.
Inflammationsresponset aktiveres af en øget mængde af lipider i blodet, og påvirker insulinsignalleringskaskaden negativt, så der sker en hæmning af både glut-4 medieret glukose transport og glukosefosforylering – således at glykolysen altså ikke aktiveres. Nedsat transport og nedsat forbrug.
Ude i vævet er det ikke nødvendigvis den klassiske randle cycle forståelse af insulinresistens der gør sig gældende, hvor pyruvatdehydrogenasen og phosphofruktokinasen hæmmes (og dermed en hæmning af glykolysen via konkurrence mellem substraterne).
– i stedet skal det forstås at det er den lipidmedierede inflammation, der er hæmmende på insulinsignalleringskaskaden, og altså ikke konkurrence om enzymer og coenzymer mellem substraterne.
Dette understreges i følgende studie, hvor man hos rotter forårsager lever insulinresistens ved infusion af frie fedtsyrer, medvirkende til en overproduktion af glukose samt hyperglykæmi.
Boden G, She P, Mozzoli M, Cheung P, Gumireddy K, Reddy P, et al. Free Fatty Acids Produce Insulin Resistance and Activate the Proinflammatory Nuclear Factor-κB Pathway in Rat Liver. Diabetes. 2005;54(12):3458-65.
——–
Tilføjelse d. 9. september 2024:
Det ser ud til at der i dette studie er brugte en fedtsyre infusion som er primært PUFA
“Liposyn II, a 20% triglyceride emulsion (Abbott Labs, Chicago, IL) was infused at 0.618 ml/h together with heparin (20 units/h). Controls received glycerol (143 μmol/h) instead of lipid/heparin. Glycerol was co-infused with insulin to match the glycerol content of Liposyn II.”
Hvis vi kigger på produktresumeet fra producenten fremgår indholdsstofferne:
“LIPOSYN® II 20% Injection contains 10% safflower oil, 10% soybean oil, 1.2% egg phosphatides,and 2.5% glycerin in Water for Injection.”.

——–
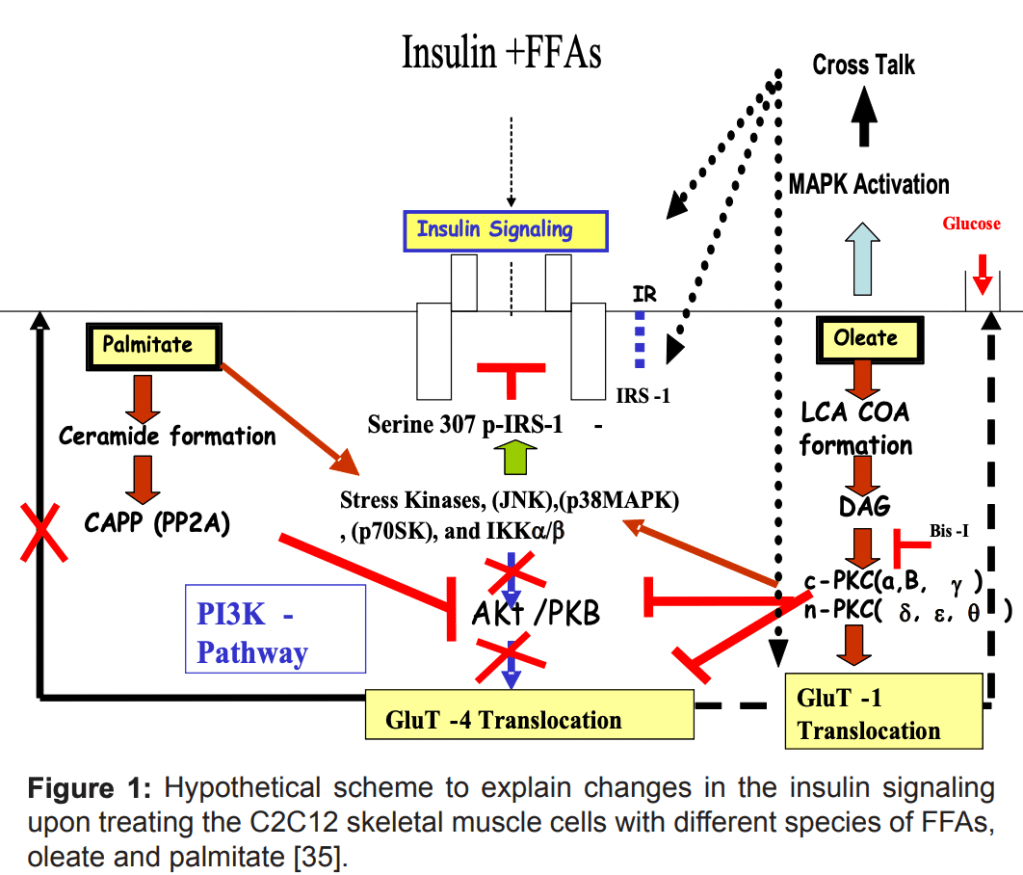
Men udover NF-kappa-beta signalleringsvejen som direkte årsagsfaktor i insulinresistens, så sker der ved akkumulering af fedtsyre produkter i cellerne, også en aktivering af Protein Kinase C/PKC, som hæmmer signalleringskaskaden.
Der sker også en fosforylering af IRS-1 som er et insulinreceptor substrat – og når det fosforyleres så hæmmes insulinsignalleringen altså.
Det ses at måde palmitat, en mættet fedt syre, kan forårsage insulinresistens, og også oleat, en PUFA/flerumættet fedtsyre kan forårsage insulinresistens, ad to forskellige veje.
Det er altså ved akkumulering af fedtsyre og dets restprodukter at insulinresistens opstår, og kan induceres af lipidinfusioner.
Så selv hos unge og raske, uden noget der minder om diabetes eller insulinresistens, kan lipidinfusioner efter få timer, forårsage insulinresistens.
For at gøre det kort:
Lipider forårsager blokade af insulinsignalleringen, bl.a. via inflammation gennem NF-kappabeta signalvejen. Acetylsalicylsyre/hæmning af inflammationskaskaden fjerner insulinresistensen ved fedtsyre infusioner. Derfor kan det altså kraftigt henlede en til at tænke at inflammation er ledet der forårsager forstyrrelser i insulinsignalleringen.
Insulinresistens er altså relateret til en øget mængde af lipider og deres restprodukter, samt den følgende inflammation.
Nedsat insulinproduktion er relateret til en over baseline forbrænding af lipider, og dertil følgende reduktiv stress med blokade af glukose metabolismen og dermed insulinfrisættelsen til følge.
Hvad kan man så gøre for at blive kureret for diabetes og insulinresistens?
– Det handler om at komme ud af reduktiv stress (elektron ophobning), øge insulinsignalleringen, og dermed øge glukosemetabolismen og sænke fedtsyre metabolismen.
En god ide kunne være at stoppe med at faste og stresse kroppen, således at man ikke aktiverer yderligere metabolsk stress.
Måske er det en god ide at reducere fedt i diæten, og især flerumættede fedtsyrer. F.eks. er der et studie hvor rotter fodres med en Svinefedt og soyaolie diæt (Høj andel flerumættede fedtsyrer, hvor NAD+/NADH ratioen formindskes – de får altså en reduktiv stress i redox balancen)
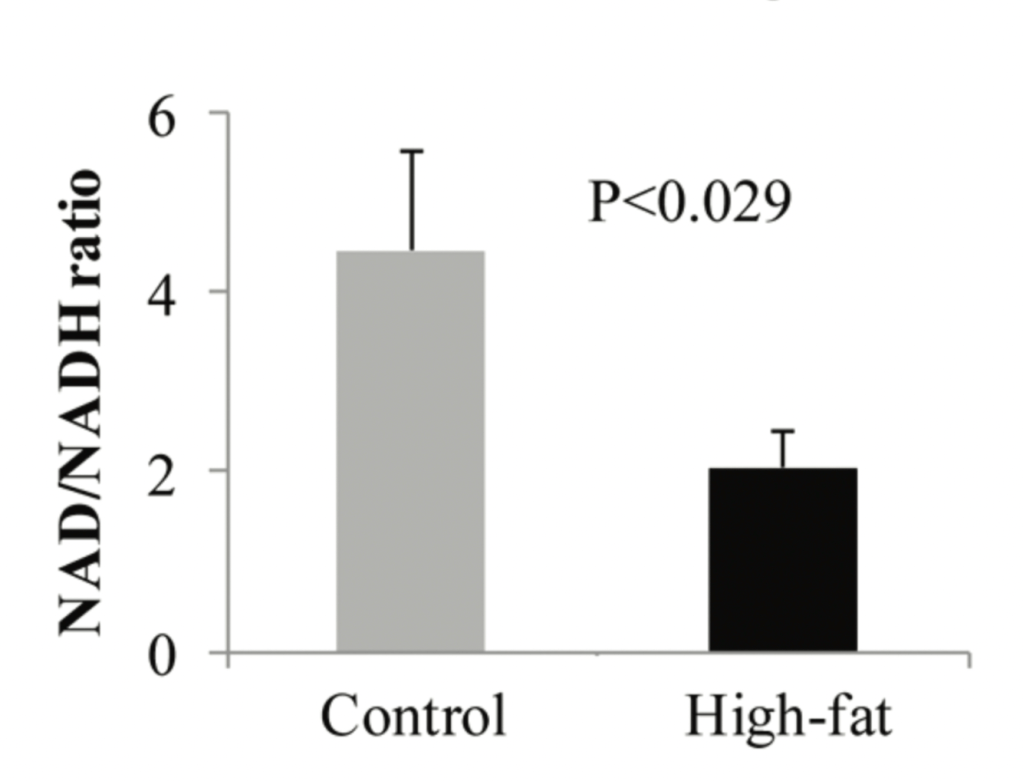
https://pubs.acs.org/doi/10.1021/pr100892r
Det har også vist sig at man kan øge ens NAD+/NADH ratio ved at øge poolen af nikotinamid/Vitamin B3 som er precursor.
https://www.sciencedirect.com/science/article/pii/S155041312030190X
Ny måde at tænke på?
Det med at det hedder sukkersyge giver ingen mening, og giver anledning til mange misforståelser om at sukker forårsager insulinresistens – når det faktisk er en en øget lipid mængde og forbrænding, der gør det svært at bruge glukose som energi.
Så for at blive kureret er handler det om at genvinde evnen til at bruge glukose som energi, så det ikke flyder over laver unødigt ravage.
Type 2 diabetes er en complex metabolsk lidelse, og vi skal i opfølgende blogindlæg se nærmere på de mange ting der spiller ind, men nu har vi i hvert fald fået argumenteret for, at noget omkring en dysreguleret glukose metabolisme i hvert fald er et stort omdrejningspunkt.


Leave a comment